This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Mice that lack the autism-linked gene neuroligin-3 show similar deficits in neuronal connections to those seen in fragile X syndrome, an inherited form of mental retardation, according to research published 13 September in Science. Restoring the gene in adolescent mice reverses the problem, suggesting a potential pathway for treatment.
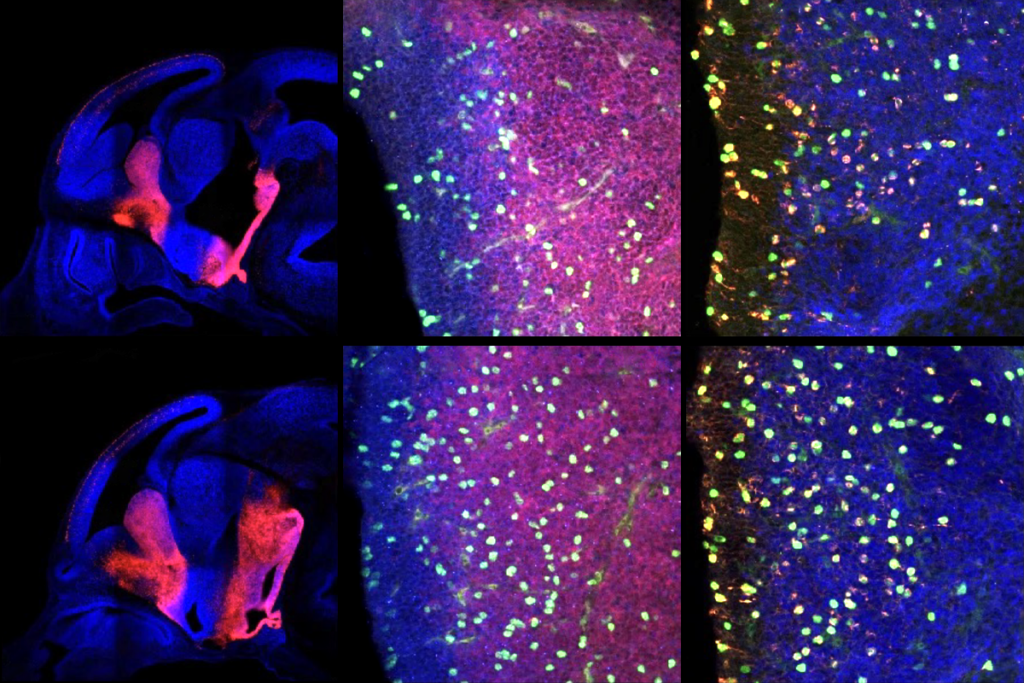
Strange synapses: Mice lacking NLGN3 develop abnormal neuronal connections (yellow) at one end of certain cells (blue) in the cerebellum.
Mice that lack the autism-linked gene neuroligin-3 (NLGN3) show similar deficits in neurons to those seen in fragile X syndrome, according to research published 13 September in Science1. Specifically, the mice have defects in neuronal plasticity, a brain cell’s ability to alter the strength of synapses, the connections between neurons.
Restoring the gene in adolescent mice reverses the problem, suggesting a potential pathway for treatment. The findings add to growing evidence that some aspects of autism may be treatable into adulthood.
“A key question with neurodevelopmental disorders like autism is to what extent can dysfunctional circuits formed during development be repaired?” says Peter Scheiffele, professor of cell biology and biological development at the University of Basel in Switzerland, and the study’s lead investigator. “We see that a substantial amount of inappropriate synapses can be rescued in the adult.”
The research also supports the idea that studying so-called syndromic forms of autism, such as fragile X, which are further along in drug development than are more common forms of autism, will shed light on the molecular underpinnings of the disorder.
“I was quite excited about this paper because it links what may be very different genetic causes of autism to perhaps similar mechanistic and therapeutic targets,” says Kimberly Huber, associate professor of neuroscience at the University of Texas Southwestern Medical Center in Dallas. That suggests that drugs being developed for fragile X might be useful for other causes of autism as well, she says.
NLGN3 is involved in the formation and maintenance of synapses. It was first linked to autism in 2003, when a mutation in the gene that produces the protein was detected in two brothers with autism2.
Single-letter mutations in NLGN3 and deletions of the entire gene have both been detected in people with autism3,4. But the effects of these mutations are complex.
Two studies in mice with a mutation that mimics the human point mutation are contradictory. One study, published in 2007, found that mice with the mutation have impaired social behavior, but a second published the following year found no differences between mice with the same mutation and controls5,6.
One of those studies also found that removing the gene completely has a different effect than the human point mutation does.
In the new study, researchers focused on the cerebellum, a part of the brain involved in motor control, language, attention and other functions. Cells in this region undergo a well-characterized type of synaptic plasticity, called long-term depression, which weakens the connections between neurons.
They found that long-term depression is impaired in mice lacking NLGN3, meaning their synapses do not weaken appropriately. “This is a hallmark dysfunction in fragile X,” says Scheiffele.
Researchers tied the plasticity deficits to too much of a molecule known as metabotropic glutamate receptor 1 (mGluR1). The molecule belongs to a family of receptors found all over the brain, and is involved in learning, memory, anxiety and other cognitive functions.
“The fact that they see enhanced levels of mGluR1 suggests that dampening mGluR signaling, which is the current hypothesis in fragile X right now, could be potentially therapeutic,” says Huber.
The mice also show signs of abnormal neuronal wiring, with synapses on parts of the neuron that don’t form synapses in control mice. This defect appears to be specific to loss of NLGN3. Mice lacking a related protein, NLGN1, have normal wiring in the cerebellum.
Restoring NLGN3 in adolescent mice, aged 30 to 60 days, reverses the deficits: The misplaced synapses disappear and levels of mGluR1 go back to normal.
Although the study focused on the cerebellum, Scheiffele says the findings are likely to be applicable to other parts of the brain. The researchers found that mGluR1 levels are also abnormal in the thalamus of the mice, but not in the hippocampus.
The researchers plan to test whether drugs can ameliorate the abnormal wiring and synaptic plasticity defects, which is essential for developing treatments. Because the plasticity problems seem to stem from an excess of mGluR1, they are collaborating with pharmaceutical companies to test compounds that block this receptor.
The approach mimics one that has proven promising for treating fragile X syndrome. Research on that disorder has focused on the mGluR5 receptor, a related molecule found extensively in the cortex. (Different mGluRs are found in different parts of the brain.)
Pharma giants Roche and Novartis are both developing drugs that block mGluR5, and Seaside therapeutics, a drug development start-up based in Cambridge, Massachusetts, is already testing its mGluR5 inhibitor in children with autism or fragile X syndrome.
Scheiffele says it may ultimately prove useful to target different types of mGluR receptors at once.
The findings also help to cement the synapse as a common target in autism. Neuroligins help maintain the molecular scaffolding at the synapse.
The new work suggests that maintaining a normal scaffolding is important for normal functioning of the mGluRs, says Huber, which is also the case in fragile X. “We are starting to form a framework linking known genetic causes of autism into a molecular model.”
1: Baudouin S.J. et al. Science Epub ahead of print (2012) Abstract
2: Jamain S. et al. Nat. Genet. 34, 27-29 (2003) PubMed
3: Sanders S.J. et al. Neuron 70, 863-885 (2011) PubMed
4: Gilman S.R. et al. Neuron 70, 898-907 (2011) PubMed
5: Chadman K.K. et al. Autism Res. 1, 147-158 (2008) PubMed
6: Tabuchi K. et al. Science 318, 71-76 (2007) PubMed