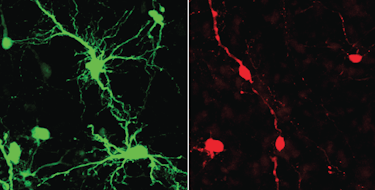
The proper regulation of neural excitatory/inhibitory (E/I) balance has been a subject of intense study in autism spectrum disorder (ASD) and other neurodevelopmental disorders. One particular focus has been the progressive increase in chloride ion extrusion from neurons as development proceeds, which is critical for the developmental “switch” in GABA function from excitatory to inhibitory. Three new studies with potential therapeutic implications shed new light on how the expression and function of KCC2 (a neuron-specific K+/Cl− cotransporter that plays an important role in this process) is regulated.
Previous in vitro studies have shown that KCC2 activity is substantially modulated by phosphorylation at two particular threonine residues. In two new papers partly supported by a SFARI Pilot Award, SFARI Investigator Kristopher Kahle and Igor Medina developed a knockin mouse model of constitutive phosphorylation at these two key threonine residues. Mice that were homozygous for these mutations died within 12 hours after birth, highlighting that precise phosphoregulation of these sites is essential for postnatal survival. By contrast, heterozygous mice were viable, allowing for an examination of subsequent neurodevelopmental effects. Among the findings was that this constitutively phosphorylated version of KCC2 prevented the normal increase in its activity during development. They associated this reduced KCC2 activity with reduced GABAergic inhibition, an enhanced E/I ratio, reduced seizure threshold, impaired social interaction and additional effects on respiration and locomotion.
In a separate study, SFARI Bridge to Independence awardee Xin Tang, together with SFARI Investigators Rudolf Jaenisch and Mriganka Sur, carried out a high-throughput screen for Food and Drug Administration (FDA)-approved drugs that might act to boost KCC2 expression in neurons derived from human embryonic stem cells. Several such compounds were identified, including those that are inhibitors of the tyrosine kinase FLT3 and the GSK-3β pathway. Of note, a few of these compounds were able to rescue phenotypes associated with Rett syndrome in both MECP2-null human neurons and Mecp2 mutant mice, including respiratory and locomotion phenotypes in the latter.
These findings give investigators new tools with which to explore KCC2 function during brain development and potentially to manipulate its activity for therapeutic benefit.

Reference(s)
Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival.
Watanabe M., Zhang J., Mansuri M.S., Duan J., Karimy J.K., Delpire E., Alper S.L., Lifton R., Fukuda A., Kahle K.
Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology.
Pisella L.I., Gaiarsa J.L., Diabira D., Zhang J., Khalilov I., Duan J., Kahle K., Medina I.
Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice
Tang X., Drotar J., Li K., Clairmont C.D., Brumm A.S., Sullins A.J., Wu H., Liu X.S., Wang J., Gray N.S., Sur M., Jaenisch R.