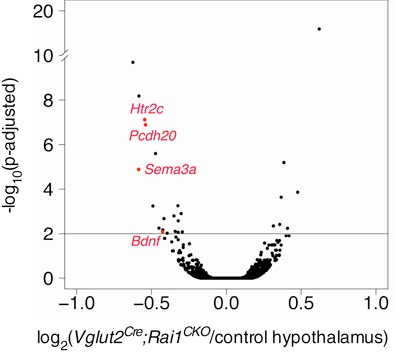
Smith-Magenis syndrome (SMS) is a neurodevelopmental disorder characterized by a diverse set of symptoms, including craniofacial abnormalities, intellectual disabilities, stereotypies, obesity and features of autism. The majority of individuals with SMS have a deletion of chromosomal region 17p11.2. Point mutations or small deletions of a single gene within this region, RAI1, are found in 10 percent of individuals with SMS. Using conditional, cell-type-specific deletions of RAI1 in mice, SFARI Investigator Liqun Luo and his colleagues provide functional insight into how RAI1 loss in distinct neuronal cell types leads to specific SMS symptoms. The researchers found that loss of RAI1 in subcortical excitatory neurons recapitulates SMS motor, learning and obesity phenotypes, with subcortical hypothalamic excitatory neurons appearing to play a particular role in obesity. The researchers also found that, at the molecular level, RAI1 enhances the transcription of genes involved in circuit assembly and neuronal communication, and links RAI1-mediated alterations in hypothalamic BDNF and HTR2C to the obesity phenotype. These findings suggest that an HTR2C agonist (which the U.S. Food and Drug Administration has already approved for the treatment of obesity) may be an effective treatment for the obesity phenotype associated with SMS.
Reference(s)
Molecular and neural functions of RAI1, the causal gene for Smith-Magenis syndrome.
Huang W., Guenthner C.J., Xu J., Nguyen T., Schwarz L.A., Wilkinson A.W., Gozani O., Chang H.Y., Shamloo M., Luo L.


