Spectrum
Spectrum is the go-to destination for the latest news and analysis about autism research and a springboard for scientists and clinicians to forge collaborations that deepen our understanding of autism.
Latest
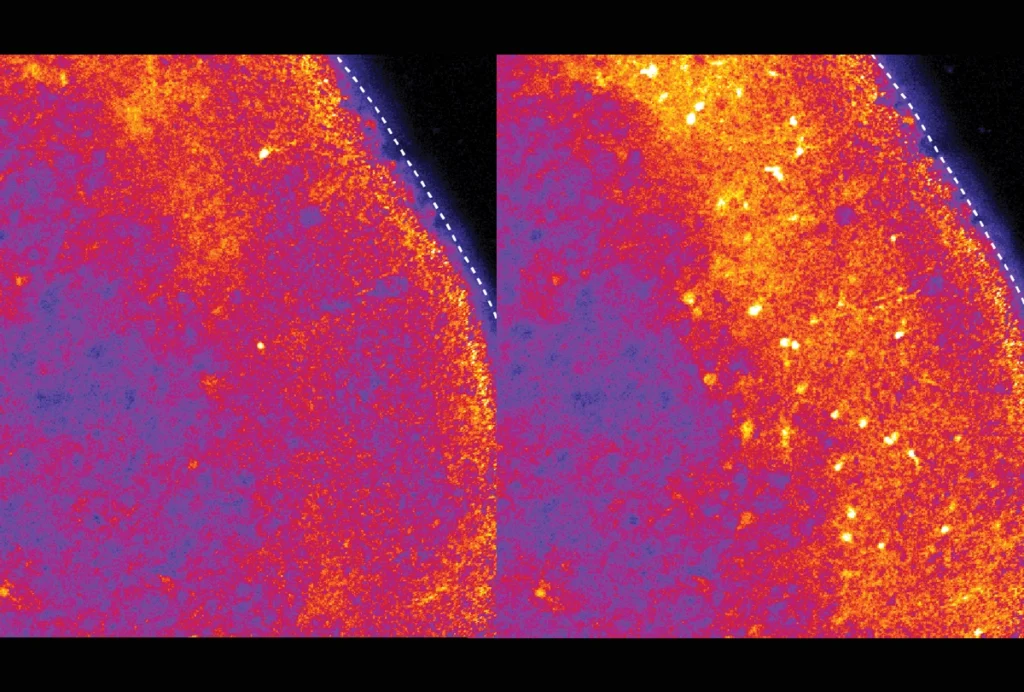
RNA drug corrects calcium signaling in chimeric model of Timothy syndrome

Reporting bias widespread in early-childhood autism intervention trials
Action potentials


A new look at walking in early childhood: Q&A with Rujuta Wilson

Common sensory response scores may miss important variations
Spectrum books

Chronicle of a Field Retold: Autism Science in Profile

Autism by the Numbers: Explaining its Apparent Rise

RNA drug corrects calcium signaling in chimeric model of Timothy syndrome
The drug, tested in rats that have human neurons, could enter clinical testing as early as next year, researchers say.
RNA drug corrects calcium signaling in chimeric model of Timothy syndrome
Reporting bias widespread in early-childhood autism intervention trials
Only 7 percent of completed registered trials were later updated with results, one of several failings identified in a new analysis.
Reporting bias widespread in early-childhood autism intervention trials
TRIO gene; left-handedness; gender diversity
Here is a roundup of autism-related news and research spotted around the web for the week of 15 April.
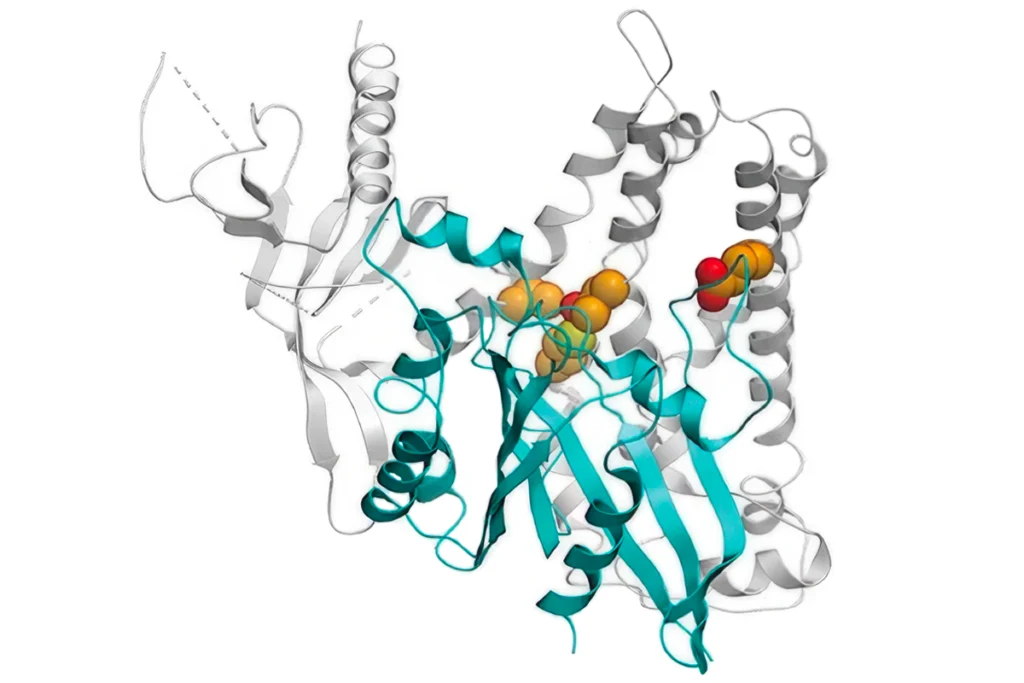
New RNA editor boasts increased versatility, safety
The “PRECISE” technique reprograms cells in a way that, unlike DNA editors, avoids potentially permanent off-target effects.
New RNA editor boasts increased versatility, safety

Acetaminophen use during pregnancy does not increase child’s chance of having autism, study finds
The link reported in prior studies likely reflects confounding factors, which sibling-matched controls in the new work address.
Acetaminophen use during pregnancy does not increase child’s chance of having autism, study finds
Explore more from The Transmitter

Crowdsourcing to curb aggression in autism: Q&A with Matthew Goodwin
To accelerate the development of real-time behavioral prediction technology, a research team is sharing data and seeking new collaborators.
Crowdsourcing to curb aggression in autism: Q&A with Matthew Goodwin
To accelerate the development of real-time behavioral prediction technology, a research team is sharing data and seeking new collaborators.

Brain connectivity and letting the data speak with Emily Finn
The Dartmouth College researcher talks about her quest to understand behavior and doing neuroscience “in the woods.”
Brain connectivity and letting the data speak with Emily Finn
The Dartmouth College researcher talks about her quest to understand behavior and doing neuroscience “in the woods.”

Carol Jennings, whose family’s genetics informed amyloid cascade hypothesis, dies at 70
Her advocacy work aided the discovery of a rare inherited form of early-onset Alzheimer’s disease and helped connect affected people with researchers.
Carol Jennings, whose family’s genetics informed amyloid cascade hypothesis, dies at 70
Her advocacy work aided the discovery of a rare inherited form of early-onset Alzheimer’s disease and helped connect affected people with researchers.