THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Two new genetic mouse models that debuted this week show that having too many or too few copies of certain genetic regions leads to an array of symptoms reminiscent of autism.
A study published yesterday in Science Translational Medicine found that mice carrying three copies of the autism candidate gene UBE3A have repetitive grooming behaviors, fewer vocalizations and little interest in social interactions1. UBE3A is located in chromosomal region 15q11-13, the most common place in the genome for genetic causes of autism.
This came on the heels of another report, published 3 October in the Proceedings of the National Academy of Sciences, on another much-talked-about region, 16p11.2. The researchers showed that mice carrying only one copy of this region have severe repetitive behaviors, larger-than-normal volumes of many brain regions and tend to die young. By contrast, those with three copies have low motor activity, smaller-than-normal volumes of the same brain areas and are relatively healthy2. These data were first presented at a Boston symposium in December.
Deletions of this segment, informally known as 16p, crop up in roughly one percent of individuals with autism. Duplications also show up in a few people with autism and, more commonly, in people with schizophrenia.
Both papers highlight what is sometimes called the ‘Goldilocks phenomenon’ of gene dosage, notes Jeremy Veenstra-VanderWeele, assistant professor of psychiatry, pediatrics and pharmacology at Vanderbilt University in Nashville, Tennessee, who was not involved in either study. “You can have too little and end up with a problem in brain development or synaptic function, or you can have too much and end up with same sort of problem.”
The new mice are useful because “we don’t know where the boundaries of these ‘genetic disorders’ really are,” he says.
Three versus two:
Researchers first linked the 15q11-13 region to autism in 19913. An estimated one to three percent of individuals with autism carry duplications of this large segment, encompassing at least 30 genes and almost always inherited from the mother.
Researchers have investigated several genes in this region, but the most famous is UBE3A. The loss of this gene causes Angelman syndrome, characterized by developmental delay, seizures, impaired speech and, intriguingly, a cheerful demeanor.”This was previously called ‘happy puppet syndrome,’ which describes the propensity for these kids with Angelman to smile a lot, to laugh a lot and to be hypersocial,” says Matthew Anderson, director of neuropathology at Beth Israel Deaconess Medical Center in Boston and lead investigator of the new UBE3A study. “It seemed to be diametrically opposite of what an autistic individual would present as.”
Individuals typically inherit two copies of each chromosome, one from the mother and one from the father. Some genetic regions within these chromosomes, such as the 15q11-13 segment, are imprinted, meaning that one of the copies is silenced.
So far, two groups have made mice carrying duplications of the entire 15q11-13 region. Others have tinkered with genes within it, including UBE3A and receptors for the neurotransmitter GABA, or gamma-aminobutyric acid4.
The new study reports the first mice carrying duplications of just the UBE3A gene, which is located within 15q11-13. The researchers engineered the mice to express either two or three copies of the gene — one or two more than normal, respectively.
Animals with three copies fail several social interaction tests, spending an equal amount of time with other mice and novel objects, whereas mice with two copies prefer to be social. Unlike healthy mice and those with two extra copies of UBE3A, those with three copies do not emit ultrasonic vocalizations in the presence of other mice.
Finally, mice with three copies of the gene show dramatic repetitive behavior in the form of excessive self-grooming, whereas those with two copies are no different than normal animals.
Unlike some other autism mouse models, these mutants showed no problems with anxiety, motor function, memory or smell. The findings suggest that UBE3A has a major role in social behavior. “It seems to be fairly limited as far as what things it impairs,” Anderson says.
Agnostic approach:
In the second new study, Alea Mills and colleagues created mice carrying either deletions or duplications of 16p11.2, which contains 27 genes. The direct comparison of these mice is important because copy number variations of this segment have been linked to a wide variety of disorders and are sometimes seen in healthy people.
“We tried to do side-by-side [comparisons] for everything,” says Mills, professor of genetics at Cold Spring Harbor Laboratory in New York. For every test, she says, she found that “it’s much worse to have too few than too many copies of this region.”
In fact, she found that half of the animals carrying the deletion die just after birth. If what they find in mice extends to humans, she says, the findings suggest that 16p11.2 deletions may cause infant mortality.
Behaviors of the mice with the deletion or duplication are often reciprocal. For example, mice with a deletion are extremely hyperactive, whereas those with a duplication are relatively lethargic. Mice with a deletion slept little, mimicking what many parents report about their children with autism, and those with a duplication slept more than normal, Mills says.
Perhaps most strikingly, the mice carrying deletions repetitively climbed across the ceiling of their cages. “The duplication mice didn’t like to crawl on the ceiling at all,” she says.
Some experts praise Mills’ study for its unusual approach to measuring behavior. The researchers used a sophisticated infrared camera system called HomeCageScan that tracks the precise movements of the mice for up to two weeks.
“We can’t interpret mouse behavior easily,” notes Veenstra-VanderWeele. The ceiling climbing behavior, for example, is “the sort of thing you wouldn’t necessarily pick up unless you took this agnostic approach.”
Still, the drawback of this method is that because the behaviors haven’t been validated on more standardized tests, it’s difficult to interpret them.
“It sounds like these guys were like Spiderman, stuck on the ceiling,” notes Andrew Holmes, chief of the Laboratory for Behavioral and Genomic Neuroscience at the National Institute on Alcohol Abuse and Alcoholism. “It’s a very interesting observation that would have led me to do more formal established tests for repetitive behavior.”
Brain measures:
Perhaps the most important contribution of these new studies is that they analyzed not only behavior, but alterations in the brain.
In the UBE3A study, the researchers showed that in the barrel cortex — the area of the mouse brain that processes sensory information from whiskers — mice carrying three copies of UBE3A have dampened transmission of glutamate. This neurotransmitter is important for excitatory signaling and has been linked to autism.
“It’s really interesting to see this signaling pathway coming up again and again,” says Holmes, whose mouse model of Williams syndrome also points to glutamate.
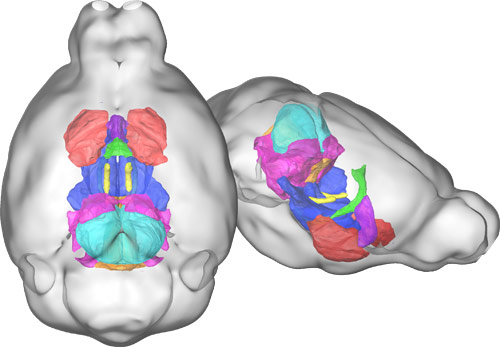
Using magnetic resonance imaging, the 16p study found that mice carrying deletions in the region have larger volumes of eight brain regions compared with controls.
“They used neuroimaging as a way to link behavioral abnormalities to what actually appears to be changed in the brain,” Veenstra-VanderWeele says. “That’s not been done in a lot of mouse models so far.”
These are only the latest two in a barrage of new mouse models of autism. In 2011 alone, researchers have described mice carrying glitches in the Timothy syndrome gene CACNA1C, SHANK3 and, just last week, CNTNAP2.
“The point of making these mice is that we can study what’s happening in the brain at a very fundamental mechanistic level,” says Ed Cook, professor of psychiatry at the University of Illinois College of Medicine in Chicago. “Now, with multiple models, we can start working on common mechanisms.”
By joining the discussion, you agree to our privacy policy.